These days, molecular biologists are dealing with lots and lots of sequences, largely due to next-gen sequencing technologies. For example, the Illumina GA2 is producing 100-200 million DNA sequences, each of 75-125 bases, per run; that works out to 20 gb of sequence data per run, not counting metadata such as names and quality scores.
Storing, referencing, and retrieving these sequences is kind of annoying. It's not so much that the files themselves are huge, but that they're in a flat file format called 'FASTA' (or FASTQ), with no way to quickly retrieve a particular sequence short of scanning through the file. This is not something you want to do on-demand or keep in-memory, no matter what BioPython says ;).
So everyone working with these sequences has evolved one way or another to deal with it. A number of different approaches have been used, but they mostly boil down to using some sort of database to either index or store the sequences and associated metadata. It won't surprise you to learn that performance varies widely; that benchmarks are few and far between; and that many people just use MySQL, even though you can predict that using something like MySQL (or really any full-blown r/w database) is going to be suboptimal. This is because FASTA and FASTQ files don't change, and the overhead associated with being able to rearrange your indices to accomodate new data is unnecessary.
A while ago -- nearly 2 years ago!? -- we started looking at various approaches for dealing with the problem of "too much sequence", because the approach we used in pygr was optimized for retrieving subsequences from a relatively small number of relatively large sequences (think human chromosomes). Alex Nolley, a CSE undergrad working in the lab, looked at a number of approaches, and settled on a simple three-level write-once-read-many scheme involving a hash table (mapping names to record IDs), another table mapping fixed-size record IDs to offsets within a database file, and a third randomly-indexed database file. He implemented this in C++ and wrote a Pyrex wrapper and a Python API, and we called it screed.
Imagine our surprise when we found out that even absent the hashing, sqlite was faster than our screed implementation! sqlite is an embedded SQL database that is quite popular and (among other things) included with Python as of Python 2.5. Yet because it's a general purpose database, it seemed unlikely to us that it would be faster than a straightforward 2-level indexed flat file scheme. And yet... it was! A cautionary tale about reimplementing something that really smart/good programmers have spent a lot of time thinking about, eh? (Thanks to James Casbon, in particular, for whacking us over the head on this issue; he wins, I lose.)
Anyway, after a frustrating few weeks trying to think of why screed would be slower than sqlite and trying some naive I/O optimizations, we abandoned our implementation and Alex reimplemented everything on top of sqlite. That is now available as the new screed.
screed can import both common forms of sequence flat file (FASTA and FASTQ), and provides a unified interface for retrieving records by the sequence name. It stores all records as single rows in a table, along with another table to describe what the rows are and what special features they have. Because all of the data is stored in a single database, screed databases are portable and you can regenerate the original file from the index.
screed is designed to be easy to install and use. Conveniently, screed is pure Python: sqlite comes with Python 2.5, and because screed stays away from sqlite extensions, no C/C++ compiler is needed. As a result installation is really easy, and since screed behaves just like a read-only dictionary, anyone -- not just pygr users -- can use it trivially. For example,
>>> import screed
>>> db = screed.read_fasta_sequences('tests/test.fa')
or
>>> db = screed.read_fastq_sequences('tests/test.fq')
and then
>>> for name in db: ... print name, db[name].sequence
will index the given database and print out all the associated sequences.
Now, because we are actually considering publishing screed (along with another new tool, which I'll post about next), I asked Alex to benchmark screed against MySQL and PostgreSQL. While sqlite is far more convenient for our purposes -- embeddable, included with Python so no installation needed, and not client-server based -- "convenience" isn't as defensible as "way faster" ;). Alex dutifully did so, and the results for accessing sequences, once they are indexed, are below:
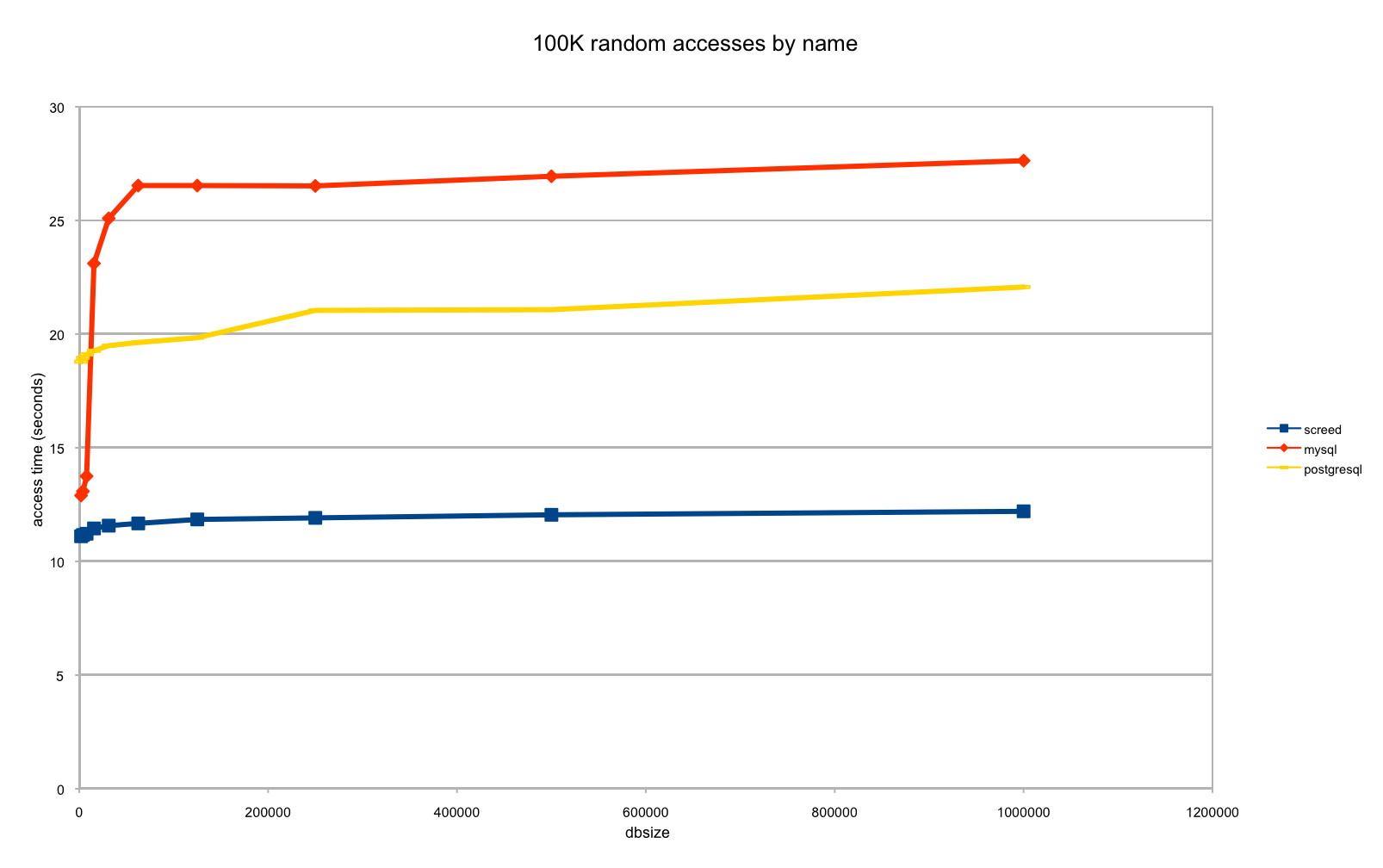
That is, sqlite/screed seems to achieve a roughly 1.8x speedup over postgresql, and 2.2x speedup over mysql, with a roughly constant retrieval time for up to a million sequences. (We've seen constant retrieval times out to 10m or so, but past that it becomes annoying to repeat benchmarks, so we'd like to get our benchmark code correct before running them.)
The benchmark code is here: screed, mysql, and postgresql. We're still working on making it easy to run for other people but you should get the basic sense of it from the code I linked to.
screed's performance is likely to improve as we streamline some of the internal screed code, although the improvements should be marginal because I/O and hashing should dominate. So, these benchmarks can be read as "underperforming Python interface wrapped around sqlite vs mysql and postgresql".
There is one disappointment with sqlite. sqlite doesn't come with any sort of compression system built-in, so even though DNA sequence is eminently compressible, we're stuck with a storage and indexing system that requires about 120% of the space of the original file.
--titus
Legacy Comments
Posted by Titus Brown on 2010-03-29 at 00:48.
Some old references: <a href="http://groups.google.com/group/pygr-d ev/browse_thread/thread/cd06c5a9f7107881">http://groups.google.com/gro up/pygr-dev/browse_thread/thread/cd06c5a9f7107881</a> <a href="http://ivory.idyll.org/blog/jan-09/lazyweb-worm-options-in- python">http://ivory.idyll.org/blog/jan-09/lazyweb-worm-options-in- python</a> sqlite does nicely in performance compared to other SQL databases as well as what we're using now, but some of these other options may also work well. Whew, there's a lot...
Posted by Titus Brown on 2010-03-29 at 00:48.
More for my own memory than anything else -- I'd be interested in trying out CDB, HDF, and pytables for some of the things I'm trying now.
Comments !