(The below post contains excerpts from Slithering your way into bioinformatics with snakemake, Hackmd Press, 2023.)
In Section 1, we introduced snakemake as a system for (efficiently and effectively) running a series of shell commands.
In Section 2, we'll explore a number of important features of snakemake. Together with Section 1, this section covers the core set of snakemake functionality that you need to know in order to effectively leverage snakemake.
After this section, you'll be well positioned to write a few workflows of your own, and then you can come back and explore more advanced features as you need them.
Chapter 4: running rules in parallel
Let's take a look at the sketch_genomes rule from the last
Snakefile entry:
rule sketch_genomes:
input:
"genomes/GCF_000017325.1.fna.gz",
"genomes/GCF_000020225.1.fna.gz",
"genomes/GCF_000021665.1.fna.gz",
output:
"GCF_000017325.1.fna.gz.sig",
"GCF_000020225.1.fna.gz.sig",
"GCF_000021665.1.fna.gz.sig"
shell: """
sourmash sketch dna -p k=31 {input} \
--name-from-first
"""
This command works fine as it is, but it is slightly awkward - because, bioinformatics being bioinformatics, we are likely to want to add more genomes into the comparison at some point, and right now each additional genome is going to have to be added to both input and output. It's not a lot of work, but it's unnecessary.
Moreover, if we add in a lot of genomes, then this step could
quickly become a bottleneck. sourmash sketch may run quickly on 10
or 20 genomes, but it will slow down if you give it 100 or 1000! (In
fact, sourmash sketch scales with the number of genomes - so it will
take 100 times longer on 100 genomes than on 1.) Is there a
way to speed that up?
Yes - we can write a rule that can be run for each genome, and then let snakemake run it in parallel for us!
Let's start by breaking this one rule into three separate rules:
rule sketch_genomes_1:
input:
"genomes/GCF_000017325.1.fna.gz",
output:
"GCF_000017325.1.fna.gz.sig",
shell: """
sourmash sketch dna -p k=31 {input} \
--name-from-first
"""
rule sketch_genomes_2:
input:
"genomes/GCF_000020225.1.fna.gz",
output:
"GCF_000020225.1.fna.gz.sig",
shell: """
sourmash sketch dna -p k=31 {input} \
--name-from-first
"""
rule sketch_genomes_3:
input:
"genomes/GCF_000021665.1.fna.gz",
output:
"GCF_000021665.1.fna.gz.sig"
shell: """
sourmash sketch dna -p k=31 {input} \
--name-from-first
"""
# rest of Snakefile here!
It's wordy, but it will work - run:
snakemake -j 1 --delete-all plot_comparison
snakemake -j 1 plot_comparison
Before we modify the file further, let's enjoy the fruits of our labor: we can now tell snakemake to run more than one rule at a time!
Try typing this:
snakemake -j 1 --delete-all plot_comparison
snakemake -j 3 plot_comparison
If you look closely, you should see that snakemake is running all three
sourmash sketch dna commands at the same time.
This is pretty cool and is one of the more powerful practical features of snakemake: once you tell snakemake what you want it to do (by specifying your desired output(s)) and give snakemake the set of recipes telling it how to do each step, snakemake will figure out the fastest way to run all the necessary steps with the resources you've given it.
In this case, we told snakemake that it could run up to three jobs at
a time, with -j 3. We could also have told it to run more jobs at a
time, but at the moment there are only three rules that can actually
be run at the same time - sketch_genomes_1, sketch_genomes_2, and
sketch_genomes_3. This is because the rule compare_genomes needs the
output of these three rules to run, and likewise plot_genomes needs
the output of compare_genomes to run. So they can't be run at the
same time as any other rules!
Chapter 5 - visualizing workflows
Let's visualize what we're doing! Here's the output of snakemake
--dag plot_comparison, visualized with the graphviz package:
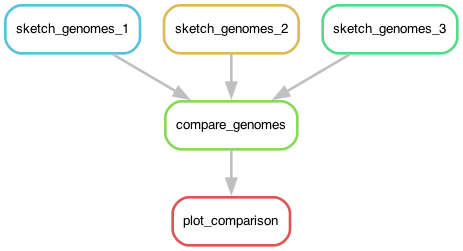
This diagram shows the relationship between the rules we've put in the
Snakefile: compare_genomes takes the output of the sketch_genome
rules as its own input, and then plot_comparison uses the output of
compare_genomes to build its own plot.
One key aspect of this graph is that it shows you where the various
rules can be run at the same time as each other because they neither
require nor are required for the others - here, the three
sketch_genome rules. That is what let us run all three simultaneously
in the previous chapter!
Note: sometimes you have to have a single rule that deals with all of
the genomes - for example, compare_genomes has to compare all the
genomes, and there's no simple way around that. But with sketch_genomes,
we do have the option of breaking the rule up!
Chapter 6 - using wildcards to make rules more generic
Let's take another look at one of those sketch_genomes_ rules:
rule sketch_genomes_1:
input:
"genomes/GCF_000017325.1.fna.gz",
output:
"GCF_000017325.1.fna.gz.sig",
shell: """
sourmash sketch dna -p k=31 {input} \
--name-from-first
"""
There's some redundancy in there - the accession GCF_000017325.1 shows up
twice. Can we do anything about that?
Yes, we can! We can use a snakemake feature called "wildcards", which will let us give snakemake a blank space to fill in automatically.
With wildcards, you signal to snakemake that a particular part of an
input or output filename is fair game for substitutions using { and }
surrounding the wildcard name. Let's create a wildcard named accession
and put it into the input and output blocks for the rule:
rule sketch_genomes_1:
input:
"genomes/{accession}.fna.gz",
output:
"{accession}.fna.gz.sig",
shell: """
sourmash sketch dna -p k=31 {input} \
--name-from-first
"""
What this does is tell snakemake that whenever you want an output file
ending with .fna.gz.sig, you should look for a file with that prefix
(the text before .fna.gz.sig) in the genomes/ directory, ending in
.fna.gz, and if it exists, use that file as the input.
(Yes, there can be multiple wildcards in a rule! We'll show you that later!)
If you go through and use the wildcards in sketch_genomes_2 and
sketch_genomes_3, you'll notice that the rules end up looking identical.
And, as it turns out, you only need (and in fact can only have) one rule -
you can now collapse the three rules into one sketch_genome rule again.
Here's the full Snakefile:
rule sketch_genome:
input:
"genomes/{accession}.fna.gz",
output:
"{accession}.fna.gz.sig",
shell: """
sourmash sketch dna -p k=31 {input} --name-from-first
"""
rule compare_genomes:
input:
"GCF_000017325.1.fna.gz.sig",
"GCF_000020225.1.fna.gz.sig",
"GCF_000021665.1.fna.gz.sig"
output:
"compare.mat"
shell: """
sourmash compare {input} -o {output}
"""
rule plot_comparison:
message: "compare all input genomes using sourmash"
input:
"compare.mat"
output:
"compare.mat.matrix.png"
shell: """
sourmash plot {input}
"""
It looks a lot like the Snakefile we started with, with the crucial difference that we are now using wildcards.
Here, unlike the situation we were in at the end of last section where
we had one rule that sketched three genomes, we now have one rule
that sketches one genome at a time, but also can be run in parallel!
So snakemake -j 3 will still work! And it will continue to work as
you add more genomes in, and increase the number of jobs you want to
run at the same time.
Before we do that, let's take another look at the workflow now -
you'll notice that it's the same shape, but looks slightly different!
Now, instead of the three rules for sketching genomes having different names,
they all have the same name but have different values for the accession wildcard!
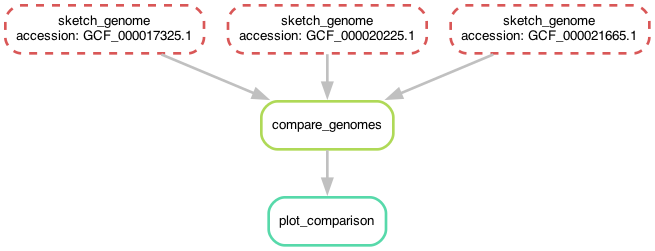
Chapter 7 - giving snakemake filenames instead of rule names
Let's add a new genome into the mix, and start by generating a sketch
file (ending in .sig) for it.
Download the RefSeq assembly file (the _genomic.fna.gz file) for GCF_008423265.1 from this NCBI link, and put it in the genomes/ subdirectory as GCF_008423265.1.fna.gz. (You can also download a saved copy with the right name from this osf.io link.)
Now, we'd like to build a sketch by running sourmash sketch dna
(via snakemake).
Do we need to add anything to the Snakefile to do this? No, no we don't!
To build a sketch for this new genome, you can just ask snakemake to make the right filename like so:
snakemake -j 1 GCF_008423265.1.fna.gz.sig
Why does this work? It works because we have a generic wildcard rule for
building .sig files from files in genomes/!
When you ask snakemake to build that filename, it looks through all the output blocks for its rules, and choose the rule with matching output - importantly, this rule can have wildcards, and if it does, it extracts the wildcard from the filename!
Warning: the sketch_genome rule has now changed!
As a side note, you can no longer ask snakemake to run the rule by its
name, sketch_genome - this is because the rule needs to fill in the
wildcard, and it can't know what {accession} should be without us
giving it the filename.
If you try running snakemake -j 1 sketch_genome, you'll get the following error:
WorkflowError: Target rules may not contain wildcards. Please specify concrete files or a rule without wildcards at the command line, or have a rule without wildcards at the very top of your workflow (e.g. the typical "rule all" which just collects all results you want to generate in the end).
This is telling you that snakemake doesn't know how to fill in the wildcard (and giving you some suggestions as to how you might do that, which we'll explore below).
In this chapter we didn't need to modify the Snakefile at all to make use of new functionality!
Chapter 8 - adding new genomes
So we've got a new genome, and we can build a sketch for it. Let's add it into our comparison, so we're building comparison matrix for four genomes, and not just three!
To add this new genome in to the comparison, all you need to do is add
the sketch into the compare_genomes input, and snakemake will
automatically locate the associated genome file and run
sketch_genome on it (as in the previous chapter), and then run
compare_genomes on it. snakemake will take care of the rest!
rule sketch_genome:
input:
"genomes/{accession}.fna.gz",
output:
"{accession}.fna.gz.sig",
shell: """
sourmash sketch dna -p k=31 {input} --name-from-first
"""
rule compare_genomes:
input:
"GCF_000017325.1.fna.gz.sig",
"GCF_000020225.1.fna.gz.sig",
"GCF_000021665.1.fna.gz.sig",
"GCF_008423265.1.fna.gz.sig",
output:
"compare.mat"
shell: """
sourmash compare {input} -o {output}
"""
rule plot_comparison:
message: "compare all input genomes using sourmash"
input:
"compare.mat"
output:
"compare.mat.matrix.png"
shell: """
sourmash plot {input}
"""
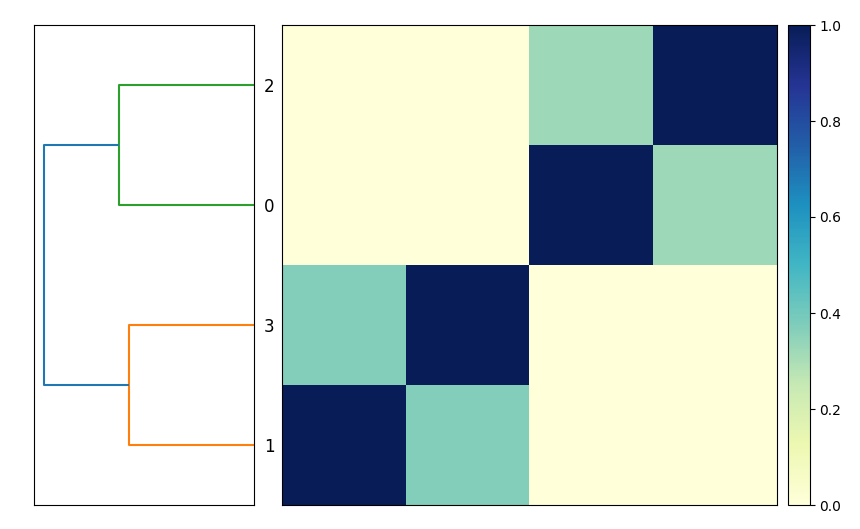
Now when you run snakemake -j 3 plot_comparison you will get a
compare.mat.matrix.png file that contains a 4x4 matrix! (See Figure.)
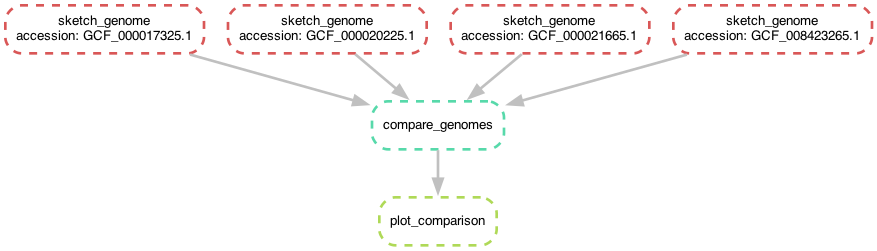
Note that the workflow diagram has now expanded to include our fourth genome, too!
Chapter 9 - using expand to make filenames
You might note that the list of files in the compare_genomes rule
all share the same suffix, and they're all built using the same rule.
Can we use that in some way?
Yes! We can use a function called expand(...) and give it a template
filename to build, and a list of values to insert into that filename.
Below, we build a list of accessions named ACCESSIONS, and then use
expand to build the list of input files of the format {acc}.fna.gz.sig
from that list, creating one filename for each value in ACCESSSIONS.
ACCESSIONS = ["GCF_000017325.1",
"GCF_000020225.1",
"GCF_000021665.1",
"GCF_008423265.1"]
rule sketch_genome:
input:
"genomes/{accession}.fna.gz",
output:
"{accession}.fna.gz.sig",
shell: """
sourmash sketch dna -p k=31 {input} --name-from-first
"""
rule compare_genomes:
input:
expand("{acc}.fna.gz.sig", acc=ACCESSIONS),
output:
"compare.mat"
shell: """
sourmash compare {input} -o {output}
"""
rule plot_comparison:
message: "compare all input genomes using sourmash"
input:
"compare.mat"
output:
"compare.mat.matrix.png"
shell: """
sourmash plot {input}
"""
While wildcards and expand use the same syntax, they do quite different
things.
expand generates a list of filenames, based on a template and a list
of values to insert into the template. It is typically used to make a
list of files that you want snakemake to create for you.
Wildcards in rules provide the rules by which one or more files will be actually created. They are recipes that say, "when you want to make a file with name that looks like THIS, you can do so from files that look like THAT, and here's what to run to make that happen.
expand tells snakemake WHAT you want to make, wildcard rules tell
snakemake HOW to make those things.
Chapter 10 - using default rules
The last change we'll make the Snakefile for this section is to add what's known as a default rule. What is this and why?
The 'why' is easier. Above, we've been careful to provide specific rule names or filenames to snakemake, because otherwise it defaults to running the first rule in the Snakefile. (There's no other way in which the order of rules in the file matters - but snakemake will try to run the first rule in the file if you don't give it a rule name or a filename on the command line.)
This is less than great, because it's one more thing to remember and to
type. In general, it's better to have what's called a "default rule"
that lets you just run snakemake -j 1 to generate the file or files you
want.
This is straightforward to do, but it involves a slightly different syntax -
a rule with only an input, and no shell or output blocks. Here's
a default rule for our Snakefile that should be put in the file as
the first rule:
rule all:
input:
"compare.mat.matrix.png"
What this rule says is, "I want the file compare.mat.matrix.png."
It doesn't give any instructions on how to do that - that's what the
rest of the rules in the file are! - and it doesn't run anything,
because it has no shell block, and nor does it create anything,
because it has no output block.
The logic here is simple, if not straightforward: this rule succeeds when that input exists.
If you place that at the top of the Snakefile, then running
snakemake -j 1 will produce compare.mat.matrix.png. You no
longer need to provide either a rule name or a filename on the command
line unless you want to do something other than generate that file,
in which case whatever you put on the command line will ignore
the rule all:.
Chapter 11 - our final Snakefile - review and discussion
Here's the final Snakefile:
ACCESSIONS = ["GCF_000017325.1",
"GCF_000020225.1",
"GCF_000021665.1",
"GCF_008423265.1"]
rule all:
input:
"compare.mat.matrix.png"
rule sketch_genome:
input:
"genomes/{accession}.fna.gz",
output:
"{accession}.fna.gz.sig",
shell: """
sourmash sketch dna -p k=31 {input} --name-from-first
"""
rule compare_genomes:
input:
expand("{acc}.fna.gz.sig", acc=ACCESSIONS),
output:
"compare.mat"
shell: """
sourmash compare {input} -o {output}
"""
rule plot_comparison:
message: "compare all input genomes using sourmash"
input:
"compare.mat"
output:
"compare.mat.matrix.png"
shell: """
sourmash plot {input}
"""
This Snakefile provides some nice features.
First, it's easy to add new genomes into the comparison - we download
the genome, name it for its accession, and add it to ACCESSIONS at the
top. Voila!
Second, we don't have to remember the names of any rules to run the whole
workflow, because the rule all: at the top provides a sensible default.
Third, it is easy to change the sketching or comparison parameters and then rerun the entire workflow from scratch - thus letting us quickly explore alternate parameters for sketching and comparisons if we so choose.
In future sections, we'll revisit this basic Snakefile from the top, and explore some of the details of rules, wildcards, and other features.
Comments !