De Bruijn graph alignment should also be useful for exploring concepts in transcriptomics/mRNAseq expression. As with variant calling graphalign can also be used to avoid the mapping step in quantification; and, again, as with the variant calling approach, we can do so by aligning our reference sequences to the graph rather than the reads to the reference sequences.
The basic concept here is that you build a (non-abundance-normalized) De Bruijn graph from the reads, and then align transcripts or genomic regions to the graph and get the k-mer counts across the alignment. This is nice because it gives you a few options for dealing with multimapping issues as well as variation across the reference. You can also make use of the variant calling code to account for certain types of genomic/transcriptomic variation and potentially address allelic bias issues.
Given the existence of Sailfish/Salmon and the recent posting of Kallisto, I don't want to be disingenuous and pretend that this is any way a novel idea! It's been clear for a long time that using De Bruijn graphs in RNAseq quantification is a worthwhile idea. Also, whenever someone uses k-mers to do something in bioinformatics, there's an overlap with De Bruijn graph concepts (...pun intended).
What we like about the graphalign code in connection with transcriptomics is that it makes a surprisingly wide array of things easy to do. By eliminating or at least downgrading the "noisiness" of queries into graphs, we can ask all sorts of questions, quickly, about read counts, graph structure, isoforms, etc. Moreover, by building the graph with error corrected reads, the counts should in theory become more accurate. (Note that this does have the potential for biasing against low-abundance isoforms because low-coverage reads can't be error corrected.)
For one simple example of the possibilities, let's compare mapping counts (bowtie2) against transcript graph counts from the graph (khmer) for a small subset of a mouse mRNAseq dataset. We measure transcript graph counts here by walking along the transcript in the graph and averaging over k-mer counts along the path. This is implicitly a multimapping approach; to get results comparable to bowtie2's default parameters (which random-map), we divide out the number of transcripts in which each k-mer appears (see count-median-norm.py, 'counts' vs 'counts2').
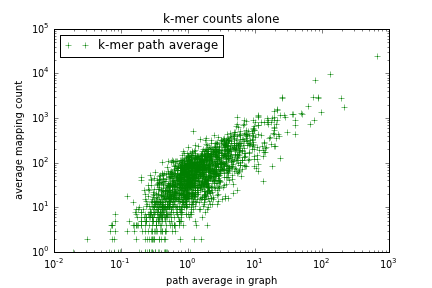
Figure 1: Dumb k-mer counting (x axis) vs dumb mapping (y axis)
This graph shows some obvious basic level of correlation, but it's not great. What happens if we use corrected mRNAseq reads (built using graphalign)?
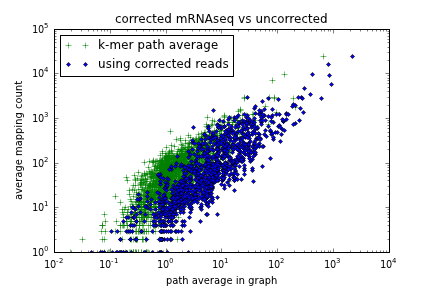
Figure 2: Dumb k-mer counting on error corrected reads (x axis) vs dumb mapping (y axis)
This looks better - the correlation is about the same, but when we inspect individual counts, they have moved further to the right, indicating (hopefully) greater sensitivity. This is to be expected - error correction is collapsing k-mers onto the paths we're traversing, increasing the abundance of each path on average.
What happens if we now align the transcripts to the graph built from the error corrected reads?
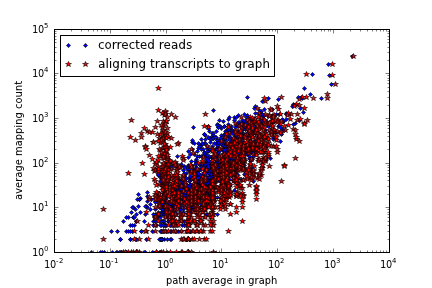
Figure 2: Graphalign path counting on error corrected reads (x axis) vs dumb mapping (y axis)
Again, we see mildly greater sensitivity, due to "correcting" transcripts that may differ only by a base or two. But we also see increased counts above the main correlation, especially above the branch of counts at x = 0 (poor graph coverage) but with high mapping coverage - what gives? Inspection reveals that these are reads with high mapping coverage but little to no graph alignment. Essentially, the graph alignment is getting trapped in a local region. There are at least two overlapping reasons for this -- first, we're using the single seed/local alignment approach (see error correction) rather than the more generous multiseed alignment, and so if the starting point for graph alignment is poorly chosen, we get trapped into a short alignment. Second, in all of these cases, the transcript isn't completely covered by reads, a common occurrence due to both low coverage data as well as incomplete transcriptomes.
In this specific case, the effect is largely due to low coverage; if you drop the coverage further, it's even more exacerbated.
Two side notes here -- first, graphalign will align to low coverage (untrusted) regions of the graph if it has to, although the algorithm will pick trusted k-mers when it can. As such it avoids the common assembler problem of only recovering high abundance paths.
And second, no one should use this code for counting. This is not even a proof of concept, but rather an attempt to see how well mapping and graph counting fit with an intentionally simplistic approach.
Isoform structure and expression
Another set of use cases worth thinking about is looking at isoform structure and expression across data sets. Currently we are somewhat at the mercy of our reference transcriptome, unless we re-run de novo assembly every time we get a new data set. Since we don't do this, for some model systems (especially emerging model organisms) isoform families may or may not correspond well to the information in the individual samples. This leads to strange-looking situations where specific transcripts have high coverage in one region and low coverage in another (see SAMmate for a good overview of this problem.)
Consider the situation where a gene with four exons, 1-2-3-4, expresses isoform 1-2-4 in tissue A, but expresses 1-3-4 in tissue B. If the transcriptome is built only from data from tissue A, then when we map reads from tissue B to the transcriptome, exon 2 will have no coverage and counts from exon 3 will (still) be missing. This can lead to poor sensitivity in detecting low-expressed genes, weird differential splicing results, and other scientific mayhem.
(Incidentally, it should be clear from this discussion that it's kind of insane to build "a transcriptome" once - what we really want do is build a graph of all relevant RNAseq data where the paths and counts are labeled with information about the source sample. If only we had a way of efficiently labeling our graphs in khmer! Alas, alack!)
With graph alignment approaches, we can short-circuit the currently common ( mapping-to-reference->summing up counts->looking at isoforms ) approach, and go directly to looking directly at counts along the transcript path. Again, this is something that Kallisto and Salmon also enable, but there's a lot of unexplored territory here.
We've implemented a simple, short script to explore this here -- see explore-isoforms-assembled.py, which correctly picks out the exon boundaries from three simulated transcripts (try running it on 'simple-mrna.fa').
Other thoughts
- these counting approaches can be used directly on metagenomes as well, for straight abundance counting as well as analysis of strain variation. This is of great interest to our lab.
- calculating differential expression on an exonic level, or at exon-exon junctions, is also an interesting direction.
References and previous work
- Kallisto is the first time I've seen paths in De Bruin graphs explicitly used for RNAseq quantification rather than assembly. Kallisto has some great discussion of where this can go in the future (allele specific expression being one very promising direction).
- There are lots of De Bruijn graph based assemblers for mRNAseq (Trinity, Oases, SOAPdenovo-Trans, and Trans-ABySS.
Appendix: Running this code
The computational results in this blog post are Rather Reproducible (TM). Please see https://github.com/dib-lab/2015-khmer-wok3-counting/blob/master/README.rst for instructions on replicating the results on a virtual machine or using a Docker container.
Comments !