At BOSC 2012, we heard a report from Richard Holland on the Pistoia Alliance Sequence Squeeze competition. I'd run across this a couple of times before -- most notably in the Quip paper -- and was interested in hearing the results.
What was the problem being tackled? To quote,
The volume of next-generation sequencing data is a big problem. Data volumes are growing rapidly as sequencing technology improves. Individual runs are providing many more reads than before, and decreasing run times mean that more data can today be generated by a single machine in one day than a single machine could have produced in the whole of 2005.
Storing millions of reads and their quality scores uncompressed is impractical, yet current compression technologies are becoming inadequate. There is a need for a new and novel method of compressing sequence reads and their quality scores in a way that preserves 100% of the information whilst achieving much-improved linear (or, even better, non-linear) compression ratios.
In the end, though, I found the results disappointing. The main conclusions?
"First, that the gzip algorithm that served as the competition baseline is actually quite sufficient for run-of-the-mill compression, and second, that it's extraordinarily difficult to make huge improvements in all three of the judged dimensions," said Nick Lynch, external liaison of the Pistoia Alliance and chair of the judging panel.
In some sense, this is pretty reassuring -- if you're compressing data, it's nice that gzip is hard to beat! Even in situations like this, where you have domain-specific data, gzip can compete reasonably well. Better compression algorithms do exist, but there's a severe time/space tradeoff.
In another sense, though, it's insane. We should be able to compress the hell out of most short-read sequencing data! Short-read sequencing data is massively redundant for two reasons. First, because to achieve sufficient coverage with shotgun sequencing you need to sample deeply on average. For example, if you have a 100x coverage genomic sample, then more than half of your content is represented more than 100 times in your data set. Second, because most of the time what you're sequencing has already been sequenced before -- if you're looking for genomic variants (in resequencing data), you expect most of the data to be identical to what you already have. That is, if you are trying to figure out how my genome is different from, say, Craig Venter's, you can rely on the fact that, as two white guys, we're probably about 99.99% identical at the genomic level. Even if you're sequencing an Australian aboriginal you can guess that our genomes would be 99.9% identical (this number is made up -- corrections welcome. Is it 99.8 instead? ;) More generally, the vast majority of sequencing is biomedical (because that's where the money is) and with only a few exceptions, the vast majority of that data is going to be from a few well-sequenced species, and (from an information content perspective) essentially the same as what everyone else is sequencing. Even if you want to keep all the single-nucleotide variants and predicted insertion-deletion variants, we're talking about a few megabytes per data set -- not hundreds.
Some short-read data is more interesting - there are some very interesting applications of sequencing, mostly in environmental, ecological, and evolutionary studies, for which the theoretical redundancy of the data sets is lower than that for straight genomic or biomedical sequencing. For example, I would venture to say that our current soil metagenomic data sets are only about 80-90% compressible; this is compared to the common genomic and RNAseq data sets, which should be (info-theoretically) between 99-99.9% compressible. (Footnote: this is my mental calculation for the ratio between an appropriately 2-bits-per-base uncompressed storage, and the maximal information theoretic lossless compression. There's some fancier term for this that I'm forgetting...)
But the main point is this -- most short-read sequencing data is crazy redundant, WAY past the few 30x maximum compression ratio achieved by even the best of the sequence squeeze participants.
So, why didn't the sequence squeeze competitors do better?
The answer is simple: FASTQ. Specifically, quality scores.
A digression: how Illumina sequencing works, kinda
So, you have some DNA. And you want to sequence it.
One very common way is to use an Illumina machine, like so:
- You fragment the DNA into molecules of length ~300-600 bases.
- You stick the DNA randomly to a 2-D surface.
- You amplify the DNA where it sits, so now you have lots of mostly identical fragments of DNA in close proximity to each other.
- You strip the DNA down to single-stranded fragments.
- Then you repeatedly add individually fluorescently colored A, C, G, and T bases to every single-stranded DNA molecule on the surface, thus reading off the next base of sequence on the opposite strand.
- As you do #5, you image the 2-D surface with a microscope.
- You take the resulting movie and simply "read" the DNA sequence present at each spot on the 2-D surface off from the twinkling little lights at each spot.
- These DNA sequences are transcoded into strings of DNA, together with a quality score that correlates with how confident the reading algorithm in #6 is about the identify of each particular character of DNA. This is all put into a FASTQ file, which is one "output" of the whole process.
You can view this as an incredibly complex but ridiculously effective way to digitize DNA into bits, and it's how most sequencing is done these days. (There are many other sequencing schemes, and no doubt this will change. But as of October 2012, Illumina is by far the most used sequencing approach in terms of volumes of data.)
Back to compression
The problem that the Sequence Squeeze participants ran into is, basically, that the terms of the context specified lossless compression -- whatever process you used had to retain all the bytes. This is really troublesome because of one main thing -- the noise inherent in the data.
First, the FASTQ format specifies a range for quality scores of between 60-90 in length, and the information that these quality scores encode is noisy. Noise is hard to encode efficiently, and the more resolution you bring to bear on the noise, the less compression you can apply. While the ultimate compression you can do on the 60-character quality-score alphabet is going to be very dependent on the frequency distribution of those 60 characters, no matter how you cut it a 60-character alphabet is much less encodable than a 4-character alphabet like DNA, especially if there's randomness in the 60-character alphabet.
Second, the DNA sequence itself is noisy, too. The error rate of Illumina is in the 0.1% to 1% range, which automatically limits the compressibility of the data. This noise essentially limits the degree to which you can use prior knowledge (like reference genomes) to compress things, because the noise is random (i.e. uncorrelated with the reference genome). But these limitations are relatively minor compared to the noise in the FASTQ format.
There are (at least) two solutions to these problems. The first is to reduce the alphabet size of the quality scores. The second (more general) solution is to allow lossy compression.
The first is obvious, and is itself a kind of lossy compression, in the sense that along with discarding the noise in the quality scores, you're also going to discard some true information. But I think the second is much more interesting.
Lossy vs lossless compression
The Wikipedia page on lossy compression is excellent. The term essentially applies to discarding information in the service of compression, where "information" is defined information theoretically to mean pretty anything non-random. Lossless compression means you can recover exactly the set of bits you had to begin with; lossy compression means that you lose some of the data. The trick with lossy compression is to pick which of the data you discard in order to minimize the effect on downstream usage.
It turns out we all use lossy compression on a daily basis -- the image and video encoding used for JPEGs and MPEGs are lossy! Most if not all lossy compression schemes allow you to specify how much information you lose, and a cool graphic demonstration of that is available here, for JPEG images. You can compress photographs a surprisingly large amount before they become indistinct!
With images, the JPEG compression algorithm is designed to keep the image as recognizable as possible. Or, to put it another way, JPEG is attempting to discard as much data as possible while retaining as much mutual information as it can between the image and the downstream application -- in this case, your visual cortex and the image recognition capabilities of your brain. If you can throw away 95% of the data and still have your lolcatz, then why not? Again, the key point for lossy compression is downstream application -- what information are you trying to keep, vs which information are you trying to discard?
For sequencing applications, there's actually a pretty solid answer to "what do we want to do with the data?" Scientists generally have one of a few immediate downstream applications in mind -- sequencing things for assembly, resequencing things to find novel variants, and resequencing things to count them -- and it is relatively straightforward to envision what lossy compression would be in these cases. Assembly is an excellent example of lossy compression, in the sense that you are trying to maximize the mutual information between your sequencing data and the predicted genome output by the assembler. For resequencing, the pattern of variants from some reference is, basically, a compressed version of the sequencing data. And, for counting applications, you can summarize counts on a per-molecule or per-region basis.
Put another way, sequencing error is the information you don't care about. You really only care about the bases that might be real. With the high error rate of sequencing, the vast majority of "information" in your data set is actually erroneous with respect to the source biological data. And this puts the Pistoia Sequence Squeeze competition into sharp relief: by insisting that software perform lossless compression, they hamstrung themselves by effectively requiring that all the error be kept around.
What do I think we should be doing?
A pipeline view
If you view sequence analysis as a pipeline -- produce data, subject data to some analysis that leads to a prediction, and use the predictions in further analyses -- then you're probably either confused or concerned right now. Sequencing and bioinformatics is a rapidly evolving field, and new sequencing technologies and analysis packages are being developed up all the time; why would we want to compress our data into a result by saving the output of, say, the GATK toolkit? That would prevent us from taking advantage of improvements to GATK, or competitors to GATK! This thinking leads to the conclusion that I bet motivated the Sequence Squeeze competition: we need to save all the raw data, in case we come up with better ways to analyze it!
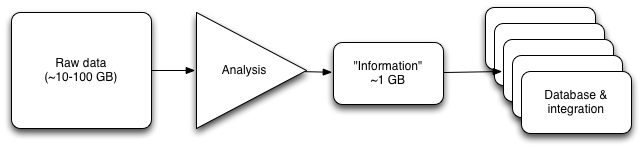
Fig 1. A somewhat abstract view of an analysis pipeline.
I'm sympathetic to the idea that we don't want to save all the output from any given analysis package. All too often we are feeding our data into a gigantic black box of an algorithm/software package/blob and we have very little idea of how it will work specifically on our data. Even when we do, we know that it's going to be making some choices with respect to sensitivity and specificity, and perhaps those will be the wrong choices.
However, given the contents of the FASTQ format, it is also pretty crazy to insist that all of that stuff is worth keeping. Complex proprietary algorithms are used to call bases and assign quality scores, and we can be reasonably certain that there are some serious heuristics (i.e. guessing) being applied; choosing to keep the exact number spit out by the sequencer is pretty arbitrary. There is little reason to believe that a 30-character quality score alphabet would be significantly worse than a 60-character quality score alphabet. Heck, in most cases, a variant call or a base call can be made with greater than 99.99% certainty; I bet you could quantize the quality score encoding down to "rock solid" and 7 shades of "less solid" without significantly affecting your base calls.
On the scale of things, I don't think we should make any one part of the pipeline sacred. We should be figuring out how to optimize the data we keep in order to meet specific limits on analysis sensitivity; and, in particular, we should be thinking about how to maximize the re-analyzability of the data in light of the specific sensitivity we need for each downstream application.
For example, if we are sequencing a single diploid individual, we can pretty confidently throw away minor variant information; if you have 100-1 allele calling at a given location your answer is pretty much guaranteed correct. If you're sequencing a population sample, e.g. a tumor, you might have more trouble picking a high threshold; heck, you might even want to keep all that data. But the choice should be made for each application.
The interesting thing is that such an "arbitrary" line in the sand has already been drawn! We don't keep the raw image files from the sequencing movies, which actually contain the maximum amount of digital information; we don't keep them because they're freakin' huge (multiple terabytes per run), and it doesn't make economic sense. It's easier and cheaper to resequence the 0.1% of samples that you need to reanalyze from scratch than it is to keep around the 99.9% of unnecessary data. It's just silly to argue that all the data is sacred, and choosing the FASTQ files as your "perfect" stage is not particularly meaningful.
So what should we keep? I bet we can compress the data down to a certain point, discarding things that are obvious errors, while still retaining all the signal and a certain amount of noise. This would still make it possible to do any downstream analysis you wanted -- just on a vastly decreased set of data.
Our digital normalization approach approximates this for assembly: it discards quite a bit of data and noise while keeping 99.999% of the information. However, diginorm discards the abundance information. Error correction, especially error correction that is tunable to retain polymorphism, would be a more sensible long term approach: it would enable lots of compression while still retaining abundance. What we're working on in my lab is technology to do this efficiently and tunably.
One of the key perspectives that I think our work (partitioning and digital normalization) brings to the table is the idea of prefiltering and eliminating known-bad data, rather than trying to guess at what is going to be good. We are trying to keep all of the information, in as generic a sense as possible, while discarding noise and scaling the problem; whether or not that information is actually used by any particular downstream analysis software is irrelevant. Another way to look at it is that we're trying to discard obvious noise and errors, rather than make specific calls about what is going to be interesting -- see Wikipedia on the LHC Computing Grid for an example of this in particle physics. This perspective is good because it gives us the ability to choose what kinds of tradeoffs we want without being unduly tied to any one downstream analysis platform.
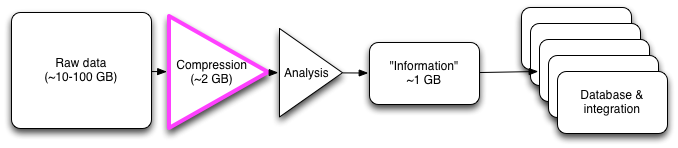
Fig 2. Inserting lossy compression into an analysis pipeline.
It's important to note that this is not a new idea by any means -- I vaguely remember reading about cram a while back. This is a package that Ewan Birney et al. are working on that does reference based compression -- exactly what I'm talking about above wrt human DNA. (Thankfully Nick Loman pointed it out to me when I asked him to review a draft of this post and so I can refer to it properly ;). Cram, like Quip, attempts to make use of assembly techniques to generate a new reference for unmappable reads, while taking the mapped reads back to the reference for reference-based compression. Perhaps most interestingly to me, they are thinking about quantizing quality scores for exactly the reasons I point out above. You can read the full paper here.
What should the next competition be?
I'd love to see an open competition like the Pistoia Sequence Squeeze contest, but focused on lossy compression, as a prefilter to one or more specific downstream packages. That is, provide a way to calculate sensitivity and specificity from the output of (say) GATK, and also measure the time and space complexity of the entire pipeline from raw data through to end analysis. You could of course do this all on the cloud, to ensure a reasonably uniform and widely available computing environment.
On the one hand, this would encourage people to shave data down to the bare minimum such that GATK with the given parameters "got it right" -- not necessarily a good outcome. But, on the other hand, it would show what the true limits were, and it would probably drive a whole bunch of research into what the various tradeoffs were and how to design such algorithms, choose parameters, and evaluate their performance.
Triage, compression, and Big Data
As one BOSC attendee told me, with the size of the data we're starting to generate, we have to figure out how to compress the data, scale or parallelize the algorithms, or discard some of it. Some data will be gold, some will be copper, and some will be dross; it should at least possible to efficiently separate much of the dross from the might-be-gold.
My real interest is in enabling the appropriate economic decisions to be made. Storage and processing time are considerations that should to be discussed and explored for data-driven biology to become less handicapped, and so far we haven't done much of that exploration. I think this could end up having serious and dramatic consequences for clinical data processing down the road, in addition to obvious research benefits.
One caveat is that there are also legal issues surrounding data retention: for example, clinical folk are apparently mandated to keep all their "raw data" (which means FASTQ, not images, apparently). I'm not prepared to take on the social and legal aspects of this proposal :). But even so, you can dump your big data to very cold storage and retain the filtered data for daily or monthly or yearly reanalysis.
Conclusions
I don't have any, really; I'll post updates from our work as they occur. I'd love to hear about other people's attempts to do this (it's not a new idea! see cram, above) and also about why you think it's a horrible idea for NGS analysis...
Special thanks to Nick Loman, Jared Simpson, and Zamin Iqbal for sanity checking and helpful references!
--titus
p.s. After writing all of this, I discovered Ewan Birney's May post on the same topic in which he basically reprises everything I said above. So, um, YEAH. The question is, can our lab put all of that on a streaming basis? Answer: yes :).
p.p.s. Jared Simpson also pointed out this very interesting paper on using the Burrows-Wheeler Transform to compress large genomic databases: Cox et al., Bioinformatics, 2012.
Comments !