We just posted a preprint, Lightweight compositional analysis of metagenomes with FracMinHash and minimum metagenome covers, Irber et al., 2022! Some day soon I'd like to write a long blog post about how this is six years in the making, part of a major intellectual endeavor in the lab that I'm incredibly excited about, yada yada yada, but for now let me just say that I think it's got some interesting ideas in it and if you're at all interested in analyzing shotgun metagenome data you should open it in a tab somewhere; a very readable HTML version is available for just that purpose.
There is a super cool figure. You should check it out!
But what I'm really here to say is this: you might see a super cool figure in the paper that looks like this:
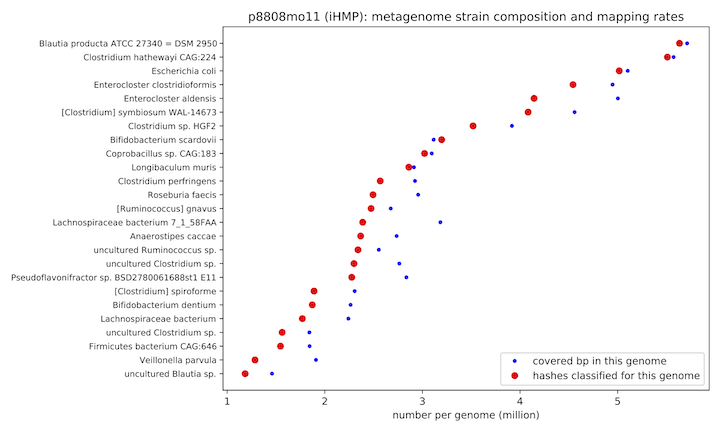
That figure is super cool in part because it tells you what microbial genomes from Genbank are present in your shotgun metagenome.
And it's even super cooler because our software figures out which genomes are present automatically, and can use all of Genbank microbial to do so!1
We're not talking taxonomic information here, BTW, where you then have to go pick a representative genome after doing an analysis that only gives you vague species-level designations. Nope, we're talking cold, hard DNA-sequence-on-the-table, genome-files-in-a-directory, automatically retrieved and analyzed for you. With mapping and everything.2
(Taxonomy is available, if you're interested in such. You can use GTDB or NCBI taxonomy as you wish. But you can just have the genomes, too!)
Where can I get this magickal software?
What, you say? How is this magic possible!?
We wrote some software! And workflows! It's called genome-grist and it's available NOW NOW NOW for the LOW LOW COST of FREE!
And (I can't stress enough how excited I am about this) it's got documentation, too!
And, for an unlimited time only, you can even integrate your own private, unpublished, cherished and hoarded genome sequences!3
<ahem>
Anyhoo. Feedback is welcome.
And yes, this is actually the software that was used to calculate the figures in the paper about the sourmash software referenced at the top. Yes, we are writing software so that we can generate figures for papers about other software. No, this will never end.
--titus
1: terms and conditions may apply: right now we can only give you Genbank as of July 2020. Sorry. We're working on it.
2: terms and conditions may apply: right now this really only works well with paired-end Illumina metagenomes.
3: And, like, your own private taxonomic classifications for your genomes, if you're into that kind of thing.
Comments !