(Happy New Year, everyone! Thanks on this blog post go out to Erich Schwarz and Taylor Reiter, for offering helpful suggestions and asking tough questions as I meandered through this work!)
Yesterday,
I posted
about using sourmash lca classify to taxonomically classify
bacterial and archaeal genomes quickly, and compared the results to
the full GTDB taxonomy. The tl;dr was that sourmash works pretty well
and returns results consistent with GTDB and GTDB-Tk, but that it
often doesn't classify as precisely as GTDB-Tk.
I was kind of expecting that at the species level, because there is a limit to the kind of precision that downsampled k-mers can achieve: the last 1-0.1% of nucleotide similarity can be a bit wobbly with sourmash (<- technical term).
But I was surprised to see the phylum and superkingdom level
limits. sourmash lca classify couldn't classify 235 genomes beyond
phylum level! What could be causing this?
Digging into a single case of imprecise classification by sourmash
I took a closer look at
GCF_001477405,
a genome tagged as Staphylococcus sciuri in Genbank. Using sourmash
lca summarize, I output a summary of the taxonomic labels of the
31-mers in this genome, downsampled at 10,000. At the phylum level, I
saw:
67.9% 199 d__Bacteria;p__Firmicutes
2.0% 6 d__Bacteria;p__Proteobacteria
which suggest that there are about 60k 31-mers in the genome that belong to genomes in the phylum Proteobacteria (they're from the Bradyrhizobium sp003020075 genome, if you're interested :).
And, because of the mechanism and thresholds by which sourmash lca
classify works, those 60k k-mers were enough to trigger confusion
about whether the genome belonged to the Firmicutes or the
Proteobacteria.
The limitations of a naive lowest common ancestor algorithm
Please indulge me in a brief digression about lowest common ancestor
approaches to classification. Per
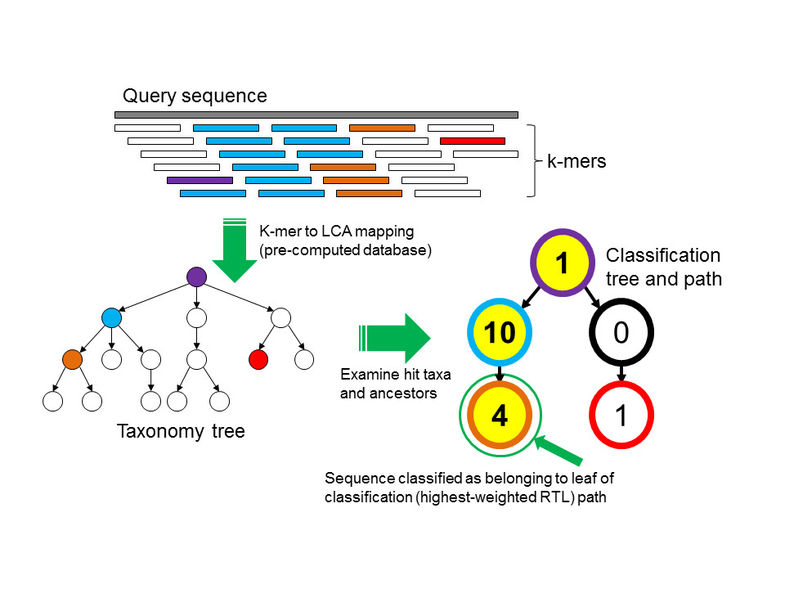 from Wood and Salzberg 2014), the algorithm for taxonomic
classification of collections of k-mers looks something like this:
from Wood and Salzberg 2014), the algorithm for taxonomic
classification of collections of k-mers looks something like this:
- Classify all k-mers individually
- Collect all the classifications into a single tree
- Compute the lowest common ancestor of all the classifications
- Assign that classification to the collection
This is the algorithm that sourmash uses, with the addition of a filtering step to remove classifications that are few in number before computing the lowest common ancestor.
(Kraken takes a more sophisticated approach than this, where it computes the highest-weighted root-to-leaf path through the taxonomy (as pictured in the above figure).)
So that's what going on with this specific genome: sourmash is doing the right thing (by our implementation of the lca algorithm), and refusing to classify the genome beyond the phylum level, because the genome has bits and pieces of firmicutes and proteobacteria. Meanwhile, GTDB is appropriately deciding that this is almost certainly a Staphylococcus sciuri, based on its marker genes.
Looking systematically at genome composition across 25k genomes
On the other hand, why the heck is 2% of this genome shared with Bradyrhizobium sp003020075?? That's a good question...
Rather than dig into this in a case by case basis, I decided to look across 25k of the GTDB genomes - these are the 25k dereplicated genomes that are part of the GTDB toolkit, and (not coincidentally) the ones in the databases that we posted on Monday. These "LCA" databases contain a dictionary of all of the k-mers in all 25k genomes, together with their taxonomic lineages - perfect!
So I devised the following algorithm:
- Gather all 51-mers in the 25k genomes
- Identify those that are "taxonomically incoherent" at the superkingdom or phylum level, by which I mean they belong to genomes in both Archaea and Bacteria, or multiple phyla within Archaea or Bacteria.
- Find pairs of genomes that belong to different phyla or superkingdoms and contain approximately 100,000 or more 51-mers in common.
This algorithm is implemented in the find-oddities.py script, if you're interested; it'll run on any sourmash LCA database file, and takes about a minute to run on the 25k genomes one.
What does the output look like? This!
cluster 0 has 2 assignments for 47 hashvals / 470000 bp
rank & lca: superkingdom d__Bacteria
Candidate genome pairs for these lineages:
cluster.pair 0.0 share 470000 bases
- GCA_003220225 (d__Bacteria;p__Methylomirabilota;c__Methylomirabilia;o__Ro\
kubacteriales;f__GWA2-73-35;g__AR12;s__AR12 sp003220225;)
- GCA_003222275 (d__Bacteria;p__Acidobacteriota;c__Vicinamibacteria;o__Fen-\
336;f__Fen-336;g__AA32;s__AA32 sp003222275;)
This is flagging two genomes, GCA_003220225 and GCA_003222275, as sharing approximately 470,000 51-mers. The output is sorted by number of shared k-mers, descending, and for the particular thresholds and parameters that I'm using, there are 21 sets of lineages in GTDB that share 100,000 or more 51-mers across the superkingdom or phylum level.
Looking at actual alignments
The big problem with the above approach is that it relies on k-mers, and downsampled k-mers at that. To dig into the actual genomes, I decided to actually do some alignments. Briefly, I gathered each pair of genomes, aligned them with nucmer, and then filtered the resulting alignments using pymummer; this is implemented in the script find-oddities-examine.py.
The resulting output is this:
cluster0.0: 557kb aln (470k 51-mers) across d__Bacteria; longest contig: 26 kb
weighted percent identity across alignments: 97.6%
skipped 79 kb of alignments in 97 alignments (< 0 bp or < 95% identity)
GCA_003222275: removed 760kb of 6756kb (11%), 154 of 628 contigs
GCA_003220225: removed 4739kb of 6031kb (79%), 95 of 167 contigs
and hopefully it's pretty self-explanatory.
The script makes some minimal attempt to "cleanse" the genomes of things that align between them, and that's what the last two lines are. But, rather than doing anything clever, I just discard any contig that has an alignment in it. This is obviously wrong in a general sense but...
...interestingly, it provides an opportunity to see that in this pair of genomes, one genome has alignments to a bunch of fragments (that's the first one), while the other has alignments throughout (the second one). The signature of this is that you can cleanly remove all of the alignments from the first genome and only get rid of 11% of the sequence, whereas 79% of the second genome goes away when you eliminate contigs with alignments.
So, in this case, it's pretty clear that the first genome is probably contaminated by sequence from the second genome.
There are other situations where it's less clear what's going on:
cluster21.0: 115kb aln (100k 51-mers) across d__Bacteria; longest contig: 1 kb
weighted percent identity across alignments: 99.2%
skipped 4 kb of alignments in 6 alignments (< 0 bp or < 95% identity)
GCF_000477555: removed 1439kb of 2775kb (52%), 163 of 207 contigs
GCF_000427295: removed 5423kb of 6292kb (86%), 88 of 202 contigs
and here we would need to dig further.
Examining potential contamination across 25k Genbank genomes
Of the 21 pairs of genomes found with the above approach, it looks like there are 11 that have cleanly isolatable contigs with taxonomically incoherent k-mers.
The most interesting one is this:
cluster 2 has 2 assignments for 25 hashvals / 250000 bp
rank & lca: superkingdom d__Bacteria
Candidate genome pairs for these lineages:
cluster.pair 2.0 share 260000 bases
- GCF_002705755 (d__Bacteria;p__Actinobacteriota;c__Actinobacteria;o__Actin\
omycetales;f__Microbacteriaceae;g__Microbacterium;s__Microbacterium esteraromat\
icum_A;)
- GCA_003265155 (d__Bacteria;p__Firmicutes;c__Bacilli;o__Mycoplasmatales;f_\
_Mycoplasmoidaceae;g__Eperythrozoon_A;s__Eperythrozoon_A wenyonii_A;)
weighted percent identity across alignments: 97.9%
skipped 45 kb of alignments in 42 alignments (< 0 bp or < 95% identity)
GCA_003265155: removed 593kb of 597kb (99%), 34 of 37 contigs
GCF_002705755: removed 534kb of 3626kb (15%), 139 of 225 contigs
here it looks like 100% of the genome of Eperythrozoon_A wenyonii_A is contained in the genome of Microbacterium esteraromaticum_A!
I'm still fine-tuning the approach but I think this is a promising way to flag Genbank genomes that are candidates for further examination.
Some concluding thoughts for today
To summarize, what we're seeing is that whole-genome approaches to taxonomic classification (either based on phylogeny of marker genes, or on whole-genome nucleotide comparisons, or both) sometimes disagree with the details of small bits of the content.
Let me hasten to add: this is a well known approach to looking at horizontal gene transfer, and the only small bits of interesting novelty here are (a) the scaling power of sourmash and (b) the large-scale application.
Concerning my own initial question: at least some of the imprecise classifications by sourmash are probably due to cross-genome shared nucleotides, some of which may be contamination (and others of which might be legitimate lateral gene transfer, plasmids, etc.) It's hard to tell without digging in further, of course!
I think it's interesting to contrast compositional approaches like the above with approaches like average nucleotide identity (ANI). ANI is a good way to do a comparison of two (or more) genomes, but it's a bulk measure that (like all bulk measures) elides details. A k-mer based approach can detect compositional commonalities between genomes, but of course has its own limitations. Using both seems like a good opportunity!
I've tried analyses like this with the Genbank taxonomy, but because that taxonomy isn't constructed using whole-genome comparisons, the results are too messy for me to look into; I'm too likely to discover that the problem is an incoherent taxonomic assignment of the whole genome, rather than a smaller portion of the genome being confused. So I'm really appreciating GTDB, which resolves a lot of these issues!
Donovan Parks made the excellent point to me in e-mail that many of the exciting new taxa in the tree of life are based on species known only from metagenome-assembled genomes, and so some contamination is not unexpected. (See also "Composite Metagenome-Assembled Genomes Reduce the Quality of Public Genome Repositories", Shaiber and Eren, 2019 and "Accurate and Complete Genomes from Metagenomes", Chen et al., bioRxiv, 2019 for some relevant discussions.) My interest, at least for the moment, is in building tools to dig into this quickly and easily; we'll see where that goes!
--titus
p.s. The full oddities-k51.txt is here, and the full oddities-k51.examine.txt is here.
p.p.s The command lines to generate the above files are in this script.
Comments !