Titus's Pragmatic Guide to Computational cis-Regulatory Analysis (for biologists)
| Date: | 7/5/06 |
|---|---|
| Revision: | 5 |
- Why cis-Regulation is Important to Your Work
- Finding cis-Regulatory Modules with Comparative Sequence Analysis
- Looking for Binding Sites
- Searching for matches to a 'consensus sequence'
- Searching with 'weighted' models of binding sites
- Searching for homotypic and heterotypic clusters
- Discovering over-represented motifs in regulatory regions
- Other computational approaches
- Testing computational predictions
- Appropriate skepticism
- There's no good solution for finding binding sites
- Conclusion
- Tools Glossary
- Further Reading and References
- Acknowledgements
- Legalese
This is an introduction to the increasingly important problem of finding and analyzing regulatory regions in animals. I'm writing it for the 2006 MBL Embryology course. It contains an overview of the basic problem, some simple and effective solutions, and general information about what doesn't seem to work. This is not an exhaustive overview. In particular, I have left out "corner cases", data and conclusions that I feel are not general.
Comments and questions are welcome; please send them to me at titus@caltech.edu.
This document will hopefully evolve as people point out mistakes and recommend new programs. You can find the latest version of this document at http://ivory.idyll.org/articles/intro-cis-reg/.
Disclaimer: This document represents my own opinions, formed over many, many years of graduate work in Eric Davidson's lab at Caltech. They are not his opinions, nor are they necessarily the opinions of anyone else in his lab or anywhere else, for that matter. This document references "evolution" and so may not be legal in Kansas. Take everything I say with a grain of salt. You have been warned.
Why cis-Regulation is Important to Your Work
A primary source of gene expression information is in the cis-regulatory region that controls the rate at which initiation of transcription occurs. This region controls not only where a gene is turned on, but when and how much. Whenever you see a tissue-specific expression pattern with an in situ, odds are that it's the direct product of a regulatory region.
What do cis-regulatory modules actually look like?
Animal cis-regulatory modules are contiguous sequences of DNA, 100bp - 1kb in size, that contain several (often many) binding sites for sequence-specific DNA-binding proteins (aka transcription factors). The order, number, and strength of the binding sites vary with the precise function executed by the module, and there is not necessarily any primary sequence identity between modules that drive genes in similar patterns.
Regulatory modules tend to regulate only one gene -- although each gene may have many regulator modules -- and modules tend to be positioned relatively close to the gene. Regulatory regions for a gene are generally contained within either the neighboring intergenic region or the introns of that gene. (There are some notable exceptions to this, of course! In particular, the smaller the genome, the more likely it is that regulatory regions will be positioned elsewhere, e.g. in the introns of nearby genes.)
I'm purposely avoiding the issues of locus control regions, long-range enhancers, and higher order chromatin structure. All three are involved in gene regulation, and are virtually impossible to investigate computationally. Luckily they seem to be relatively rare -- or perhaps we just haven't found them yet...
What do binding sites actually look like?
Binding sites are 6-30 bp regions of DNA that are recognized by transcription factors. There are no constraints on what can be a binding site; even protein-coding sequences sometimes contain binding sites.
The single most notable feature of binding sites is that all of the known binding sites for a given transcription factor can almost always be aligned without any gaps. [1] This does not mean, however, that binding sites can be recognized easily: the suite of possible binding sites for a particular transcription factor may all share key individual bases, but in general binding sites are highly degenerate and don't share most of their sequence. This means that binding sites for the same transcription factor cannot be determined by strict sequence identity.
| [1] | Transcription factors with multiple DNA binding domains, e.g. many zinc finger factors, are notable exceptions to this: they tend to bind to multiple short binding sites with flexible inter-site spacing. |
Why is this a hard problem?
Finding regulatory regions and binding sites is a hard problem, even if you're looking in a completely sequenced genome.
In rough order of degree, the problems are:
- We still know relatively little about the in vivo binding of most transcription factors. Even in vitro data is hard to come by. Moreover, Most transcription factor data describes the part of the binding site that is necessary for binding, but does not address sufficiency.
- Genomes are very big. This leads to many false positives when searching with too general a search technique.
- Regulatory modules contain many sites that work together, and the boundary of a module can be difficult to find.
- cis-regulatory modules that have similar function often do not share much detectable sequence identity.
- Regulatory regions and binding sites both can evolve neutrally, for two reasons. Binding sites in regulatory regions are not always closely spaced, and the regions between sites are often non-functional. Also, binding sites themselves can vary substantially while remaining functional, because they can be highly degenerate. See the work by Ludwig et al. in four Drosophila species for an example of compensatory site loss and gain.
Taken together, these problems make it very difficult to leverage existing data to find regulatory regions computationally.
Finding cis-Regulatory Modules with Comparative Sequence Analysis
The only fairly reliable technique for systematically finding regulatory regions is comparative sequence analysis. This technique compares evolutionarily related (homologous) genomic regions and picks out shared sequence. Usually this shared sequence is in the form of large colinear blocks that can be aligned with strong sequence similarity, suggesting that regulatory regions rarely invert, move, or are shuffled.
When it works, it works really well
We (the Davidson Lab) have used comparative sequence analysis systematically, on 10-20 genes, with great success. Other labs have used comparative sequence analysis on C. elegans and vertebrate genomes, also successfully.
Nearly every tool built to do comparative sequence analysis works pretty well, although the target audiences vary. Some tools are intended for already-sequenced genomes, while others are intended for people doing directed sequencing of their unsequenced genomes.
There is also some interesting science in there. Look at the following image:
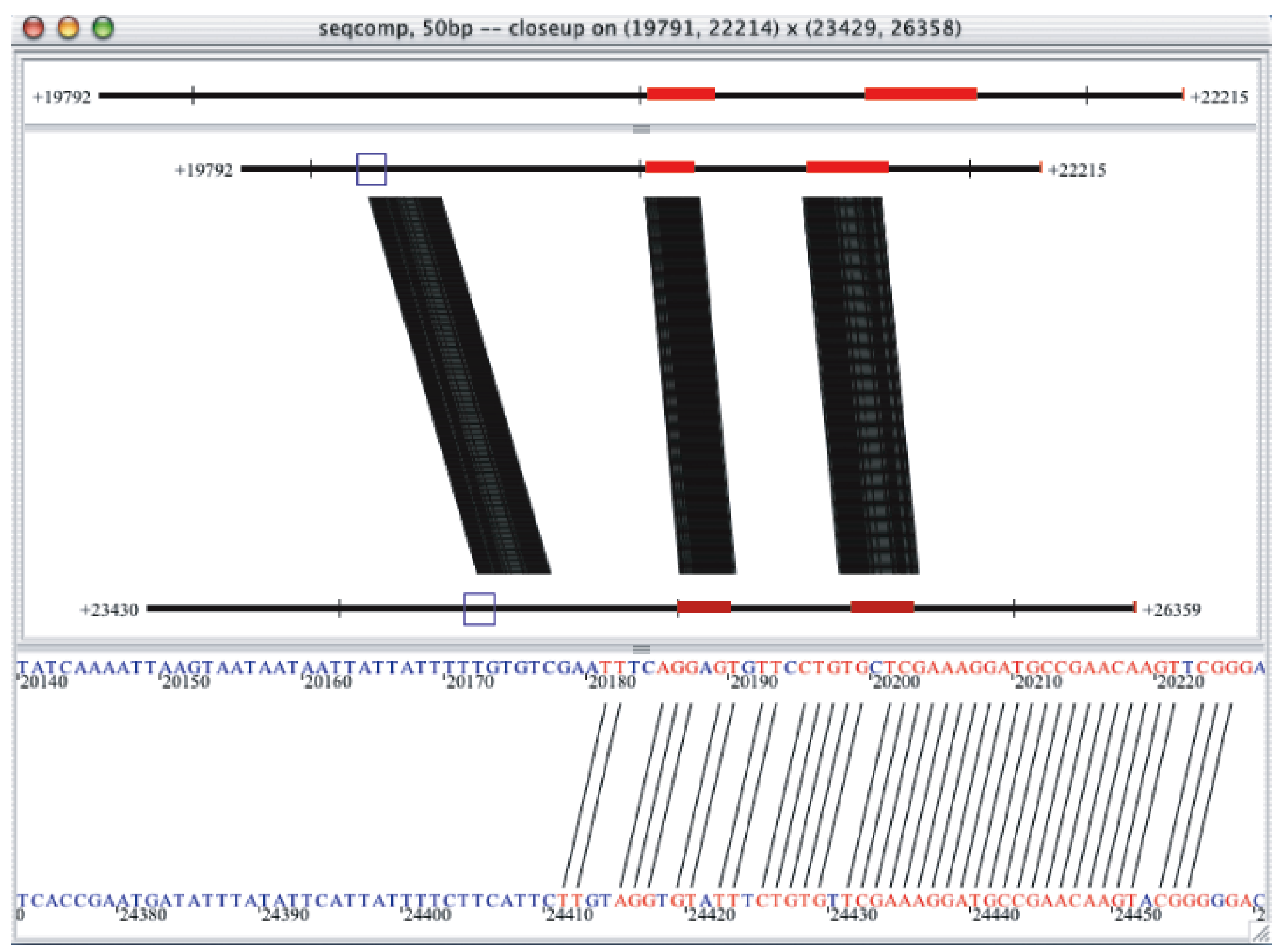
The left-most conserved block is in non-coding sequence, and it is a verified regulatory region (for the gcm gene in S. purpuratus). Note that even the left-most edge of this block is very well conserved, at the 80%/50bp level, with no insertions or deletions -- just a few point mutations. This is a general feature of regulatory regions in most animals (although see the note about Drosophila, below). The reason it's strange is that most regulatory regions should be fairly mutable -- the binding sites are dense, but not overlapping. It should be possible to hit regulatory regions with point mutations without causing any problem at all.
So, if regulatory regions should be mutable, but in practice don't get changed that quickly, what's going on?
To figure this out, Andy Cameron et al. took several sea urchin genomic regions where the functional regions had been mapped experimentally. They then looked at what happened to both functional and nonfunctional regions across various divergence times. What they discovered was that while the functional sequence was completely preserved, the non-functional sequence changed fairly fast, albeit not by point mutations but rather by insertions and deletions (indels). What may actually happening is that regulatory regions are refractory to the indels that compose the majority of mutations to inherited DNA.
I think this is pretty cool, myself, and it may explain why comparative sequence analysis works so well.
When doesn't it work?
There are several situations where comparative sequence analysis doesn't seem to work.
The first is across animals separated by large distances: only in very rare circumstances do animals from different phyla share much non-coding sequence. Moreover, even between animals within the same phyla -- for example, sea urchins and starfish, both echinoderms -- there seems to be a substantial amount of neutral divergence, where genes that are regulated by the same factors in both systems do not have similar regulatory sequence. A rough guideline is as follows:
- within the vertebrates, chick/human works very well. zebrafish and fugu have diverged a fair bit from the mammals but comparisons will still find core elements.
- for echinoderms, try to stay within 50-100 my of divergence.
- for worms, C. elegans and C. briggsae are a bit too far apart to catch everything. Some of the new genomes that will soon be available might be better.
- the two Ciona genomes seem to compare well.
Let me know if you have positive experiences with other genome pairs, please!
The second situation where comparative sequence analysis may not work is functional divergence: if two homologous genes are not regulated in the same way, then they probably will not have common regulatory modules! Note that homology of the genes is not sufficient to yield non-coding conservation; you must also hypothesize that the regulatory regions themselves have conserved function. (This may be one reason why analyzing paralogous non-coding sequence doesn't work so well; paralogous regulatory regions may diverge quickly.)
The third situation in which comparative sequence analysis apparently doesn't work is in the Drosophilids. Even with many sequenced Drosophila genomes, comparative sequence analysis has not yet been very successful. The reason for this is not clear.
What tools should I use?
In rough order of popularity:
VISTA
VISTA is a tool that does a global alignment of two large genomic regions, and then runs a window along the alignment and highlights those segments with strong identity.
VISTA will not find conserved regions that are out of order or inverted. This seems to be a minor concern, however, because in practice evolution doesn't seem to shuffle or invert regulatory regions.
PipMaker
PipMaker is a display tool layered on top of a BLAST algorithm, i.e. a local alignment algorithm. blastz, the program used to generate the local alignments, will find all points of similarity between any two regions. PipMaker can thus be used to find conserved regions that have been inverted or shuffled.
FamilyRelationsII
FRII is a display tool that can display pretty much any kind of comparative analysis. The most commonly used analysis program is seqcomp, which does an all-by-all comparison (i.e. a dotplot) using a window size and minimum threshold specified by the user.
I've been told that FRII is by far the easiest to use of the available programs. I am, however, the author of FRII -- so I might be biased. You can find out for yourself by running through the tutorial at
FamilyRelations and the associated Web server, Cartwheel, were built explicitly to let us use sequence from of directed BAC sequencing in comparative analyses. If you have a sequence from an unsequenced genome, or "rough-draft" sequence that isn't annotated, FRII should work well.
In practice, any of these programs will work. All of them will let you annotate your genomic sequence with the positions of exons and other features. Pick whichever one suits your working style best.
Looking for Binding Sites
There is no tried-and-true computational method for finding individual binding sites, either in known regulatory sequence or on a whole-genome scale. Period.
Even worse than there being no actual method, you can waste a whole heck of a lot of time trying out approaches that seem obvious, or intuitively match your expectations.
To see what I mean, let's go through a typical scenario. Let's suppose that you have a functional chunk of DNA, i.e. a functional regulatory region. You know it's functional because you tested it experimentally. But now you want to verify the transcriptional regulatory inputs; let's say that you expect a GATA factor to be directly involved in the regulation.
Searching for matches to a 'consensus sequence'
The first thing everybody does is search for a match to a "consensus sequence". This is a sequence that matches to all of the known binding sites. In the case of GATA factors, the consensus binding sequence is WGATAR, equivalent to "A or T, followed by GATA, followed by an A or G".
This approach doesn't work well, for at least two different reasons.
The first reason: it is virtually guaranteed that you will find matches to a short motif within any sequence. If you assume that A/T occurs with the same frequency as G/C, then WGATAR will occur once every half kb of sequence; we'll talk about how to calculate this below, but essentially any short (6-8bp) motif has a dismayingly high probability of occurring within any genomic region. This probability only increases as the specificity of the motif decreases, i.e. 'WGATAR' occurs four times more frequently than 'AGATAG'.
The second reason is that consensus sequences rarely accurately represent the actual binding of the transcription factor in question. There are, in turn, many reasons for this; one (discussed in the next section) is that transcription factors care much more about some nucleotide positions in their binding site than about others. Another problem is that a single outlier motif can drastically decrease the specificity of your consensus sequence, even if it's a motif that binds only weakly; for example, if you generated the WGATAR consensus sequence from these two sites:
AGATAG GGATAA
and then found a third, weaker site:
CGATAG
you would generate the consensus sequence 'VGATAR', which matches 25% more sites than 'WGATAR' -- that's a loss of approximately a 3rd of the consensus specificity -- for the sake of encompassing only one additional known site.
The most serious reason -- one that I believe pervasively skews our perceptions of binding sites in general -- is that most binding sites are determined by experiments that target only the core of the binding site. WGATAR was determined to be necessary for binding by lining up many known GATA-factor binding sites and then mutating the bases at the core of the alignment. This does not establish sufficiency, only necessity. Nobody really believes that a WGATAR motif in the middle of random sequence is sufficient for binding of GATA factors, but this distinction is not often made clearly.
In the next section, we provide a simple (simplistic?) way to deal with the first problem, that some base positions are preferred over others.
Searching with 'weighted' models of binding sites
After consensus sequences, the next best approach to try is to use a matrix of weights to describe preferences for specific bases. This approach comes with multiple names: "energy operator", "position weight matrix" (or PWM), "position-specific scoring matrix" (PSSM), or "position-specific frequency matrix" (PSFM). Rest assured that they all mean essentially the same thing; I'll use "position-weight matrix", or PWMs, in this text.
PWMs are matrix representations of motifs that allow for flexible matching to motifs. Both exact sites and consensus sites can be represented as PWMs; for example, the consensus sequence for GATA factors, 'WGATAR', can be represented in matrix form like this:
( A C G T ) 1. 1 0 0 1 2. 0 0 1 0 3. 1 0 0 0 4. 0 0 0 1 5. 1 0 0 0 6. 1 0 1 0
You may have seen PWMs represented as "sequence logos" containing DNA characters sized relative to their importance. For example, this matrix looks like this in sequence logo form:
So, in general, a PWM is a 4xN matrix, where N is the length of the site for which you're searching. A 6-base PWM like the one above can score any 6-base site; the score is generated by looking up the number for each base in each position. So, under the above matrix, 'AGATAA' would have a score of 6, while 'AGATAC' would have a score of 5. The higher the score, the better the match. To use this PWM to search a sequence for binding sites, you would simply score every N-mer in the sequence and then pick those above a certain threshold value.
PWMs are particularly useful when you want to bias the results of the motif search towards motifs that have a particular base in a particular position. For example, suppose you knew that GATA factors actually preferred an 'A' in the first position; you could represent this by making the following change:
( A C G T ) 1. 2 0 0 1 2. 0 0 1 0 3. 1 0 0 0 4. 0 0 0 1 5. 1 0 0 0 6. 1 0 1 0
Then motifs that match the consensus and have an A in the first position would have a score of 7, while consensus-matching motifs with a 'T' in the first position would only have a score of 6.
Now, the goal of PWMs is to provide a way to generalize from a list of known sequences without overgeneralizing as you do with consensus sequences. Suppose you have the following list of known GATA-binding sites:
AGATAG TGATAA AGATAA TGATAG AGATAG AGATAA AGATAA CGATAA
Just scanning down the list of sites, you can see that 'C' is a very rare occurrence in the first position, while 'A' is very common in the last position. One way to write a PWM for this set of sites is:
( A C G T ) 1. 5 1 0 2 2. 0 0 7 0 3. 7 0 0 0 4. 0 0 0 7 5. 7 0 0 0 6. 5 0 2 0
While the consensus sequence for these sites would still be 'VGATAR', because of the lone 'C' occurring in the first position, the sequence logo representation of the PWM tells a different story:
Here we see that an 'A' in the first position is much more important for a high-scoring GATA binding site than a 'C' or a 'T', which matches our intuition from looking down the list of sites.
So PWMs are basically ways of scoring motifs more flexibly; they give you the option of grading motifs differentially based on specific nucleotides in specific positions.
Of course, there are problems. (There are always problems.) Specifically, there are two problems with using PWMs. The first problem is that you must choose a threshold or cutoff at which you will no longer accept sites as being real. An N-length PWM will rank all N-mers in the genome by similarity to the input sites - but it will not tell you where the similarity becomes so low as to be unacceptable. That must be decided by the user: too high, and you will not generalize enough from your list of known sites; too low, and you will admit too many spurious sites ("false positives").
The second problem is that usually the list of known binding sites for a particular transcription factor is relatively small, which leads to a poor estimate of the "true" PWM.
Combined, these two issues mean that using PWMs subjects you to too many false positives, which in turn means that testing the predictions is both time consuming and wasteful.
A third (relatively minor) problem is that of base interdependence. It may be that one position in a binding site influences the transcription factor's specific preference for another position; there's certainly nothing biochemical preventing this! Were this alteration significant for binding, it would severely throw off the PWM prediction. However, it's a minor problem because in practice most binding sites seem to obey base independence. (In addition, we never really have enough data to train a more complicated matrix model that would take these interdependencies into account, either.)
If you want to do a PWM search, here are a few tools. The JASPAR site contains PWMs built from SELEX experiments; you can scan a piece of DNA for good matches to JASPAR binding sites at CONSITE. There are also individual Web sites that you can use to scan model organism genomes for binding sites using an alignment generated from known sites; e.g. see CisOrtho for a tool to scan C. elegans and C. briggsae. The bottom line, however, is that using PWMs to look for binding sites is still a bit of a losing battle, and you might be better off doing experiments instead.
Searching for homotypic and heterotypic clusters
Most successful binding site searches have looked for clusters of known binding sites; it makes sense to do this for binding sites that are known to work together, and especially when you're looking for sites that bind the same transcription factor cooperatively.
This has been done successfuly with both Dorsal and Su(H) binding sites in arthropods. Check out the WormEnhancer and FlyEnhancer tools.
There are two problems with this approach. First, it's not clear that it works well in the larger genomes -- both worm and fly have relatively small genomes, and the false positive rate of these tools tends to increase linearly with genome size. The second problem is simply that you have to have a pretty good idea of what you're looking for, which defeats the purpose of using computational tools!
Discovering over-represented motifs in regulatory regions
One common approach to motif discovery is to look for motifs that are in common between co-regulated or conserved DNA sequences. Tools like MEME and AlignACE build PWMs by scanning input sequences for over-represented subsequences.
This approach rarely works well unless you have a large set of sequences containing a fairly similar set of binding sites. This is largely because of the degeneracy problem: many perfectly valid binding sites may look quite distinct. Also, sensitive algorithms may pick up binding sites for architectural proteins, basal promoter sequences, repeat elements, or pretty much any non-random sequence -- of which there is quite a bit in genomes!
Other computational approaches
Essentially any biological information you have can help make a computational search more specific by cutting down on false positives. Sometimes transcription factors are known to bind cooperatively with stereotyped spacing and orientation; then you only need to look for that specific pattern of sites.
This has worked fairly well in a few specific cases; see Markstein et al. (2004).
Testing computational predictions
There are several ways to test computational binding site predictions, but not all are equal.
The best is to use chromatin immunoprecipitation (ChIP) to test whether or not the predicted binding site is being bound in vivo. This requires an antibody to the transcription factor in question, as well as a negative control -- a sequence that you know isn't bound by the transcription factor. If you have the materials, ChIP is the best technique to use, because it can verify both transcription factor identity and binding site.
A more common test is to take an already-functional cis-regulatory construct containing the binding site, and then remove the site using site-directed mutagenesis. However, the result can be difficult to interpret, especially if the effect of the site loss is quantitative only and not spatial. You may also run into problems with redundancy or partial effects if e.g. the binding site is one of several.
Combining site-directed mutagenesis with an upstream perturbation -- e.g. knocking down the putative binding factor with a morpholino -- is an excellent way to verify the identity of the binding factor.
Another type of binding site test that is common but isn't necessarily very informative is the gel shift. The problem with in vitro assays like this is that they will only verify that the transcription factor binds to the site under the in vitro conditions you're using. Unfortunately, in vivo conditions can vary significantly from the in vitro conditions: there may be additional binding partners, or similar (and perhaps competing) factors, or different accessibility to the site because of higher-order chromatin organization. Because of this I generally regard gel shifts as merely indicative, but by no means definitive.
One type of test that usually doesn't work is to attach a multimer of the predicted binding site to a basal promoter and reporter gene. This will only yield results if the binding site is sufficient to drive expression in that situation; such binding sites are rare. Moreover, multimerized sites may be in a poor configuration for actual binding. So don't do this!
While I'm generally skeptical of computational predictions, it is increasingly useful to validate computational predictions computationally. For example, if your binding site is in a region conserved among nearby species, or all of the predicted binding sites for your neurogenic transcription factor are within 1 kb of neural genes, the binding site(s) may be worth testing. For the moment I don't think that there are generally useful techniques for computational binding site validation other than looking at conservation. Howeve, as our tool set and data sets increase, computational validation will become increasingly useful.
Appropriate skepticism
I find that many biologists are inappropriately positive about computational results, especially when they match expectations! I can't stress enough the need to be skeptical about motif matching results; in particular, be sure to do negative controls. Suppose you're looking at a small region that you know to be functional, and you find a good-looking binding site. What happens if you search nearby regions with the same parameters? Do you get tons of binding sites, or only one or two? If you get lots of binding sites in regions outside your known functional region, it suggests that your results are not very specific.
I think that, in general, it's not worth doing blind motif searches. Unless you have a small set of definite targets for which to search, don't go the computational route.
Here is are some binding site predictions for a 500 bp piece of DNA.

The predictions were generated by CONSITE using the JASPAR database of transcription factor binding sites. Looks like a pretty interesting piece of DNA, eh? Well, unfortunately, the sequence was generated randomly using a computer, so unless I'm really unlucky it's completely nonfunctional. (No, I haven't tested it.) That means these binding site hits must be completely spurious.
This illustrates the central problem with doing blind motif searches quite well: too many hits. No biologist wants to spend their time doing experiments to test predictions that are 90% wrong.
There's no good solution for finding binding sites
Computationally, there's no single slam-dunk solution. There are relatively few tools that are usable; many of the tools require information that you probably don't have; and even then, the conclusions gleaned from computation need to be examined with a skeptical eye.
One bright note is that when several de novo computational prediction tools agree, it may be a trustworthy prediction. See Tompa et al., 2005.
Experimentally, the options for finding binding sites are only a bit better. Regulatory module function is often encoded in complex interactions between multiple binding sites; this makes serial deletion of regulatory regions a dangerous way to find binding sites, because you cannot reach simple conclusions without knowing what you're deleting. Chromatin immunoprecipitation works well for situations where you can get sufficient material -- transcription factors tend to be rather rare, especially in early embryogenesis. Better microarrays (for whole genome site mapping) and better ChIP protocols are fixing this, however. So far the best way to experimentally find binding sites has been to systematically probe the regulatory region by gel shifts with nuclear extract, but this is slow and technically challenging.
A middle ground that we're exploring in the Davidson Lab is to "guess": take the known regulatory module, find likely upstream factors, perturb them to verify that they're actually upstream, and then search for their likely binding site(s) using the techniques above. This technique works well in conjunction with work on gene regulatory networks.
Conclusion
Computational analysis of cis-regulatory regions is a very active area of research, but so far it's much easier to find cis-regulatory modules than it is to determine binding sites.
It sounds trite, but the most important aspect of using computational tools is to do the proper controls. By this I don't mean experimental controls, but rather computational controls: e.g. check to make sure that the specific-looking binding site arrangement you found doesn't occur throughout the genome.
Also, be aware of the evolutionary relationship of your sequences. Sequence divergence can give you a very different comparative signature from functional convergence. Sequence divergence may lead to regulatory modules that are highly conserved partly as a side-effect, making tools like VISTA, PipMaker and FamilyRelations the appropriate technology to use. Functional convergence means that similar binding sites may be present, but there is unlikely to be significant sequence similarity between two functionally similar modules unless they are also evolutionarily related. This means that more sensitive (and potentially more error-prone) tools must be used.
Tools Glossary
AlignACE (http://atlas.med.harvard.edu/cgi-bin/alignace.pl)
AlignACE is one of the original "Gibbs sampling" algorithms for novel motif discovery. It works really well in yeast, but you need to pick your sequences carefully. It doesn't have a graphical interface so it's not usable by bench biologists at the moment.
Cistematic (http://cistematic.caltech.edu/)
Cistematic is a suite of programs by a fellow Caltech bioinformatics dude, Ali Mortazavi. Cistematic seeks to integrate binding site searches with conservation information. He says it works well, and it seems to have done a great job in human/mouse motif search. It's not particularly usable by bench biologists at the moment, however.
ClusterBuster (http://zlab.bu.edu/cluster-buster/)
ClusterBuster does combinatorial binding site search in medium-sized chunks of DNA. You need to know the binding sites to enter, in general. I have heard good things about it from biologists.
CONSITE (http://www.phylofoot.org/consite/)
CONSITE looks for binding sites in conserved sequence and can also scan sequences with sites from the JASPAR database of known binding sites.
FamilyRelations/Cartwheel (http://family.caltech.edu/tutorial/)
My program for comparative sequence analysis. I like it. I hear that it works pretty well in many species, and it has a nice tutorial.
FlyEnhancer (http://flyenhancer.org/Main)
FlyEnhancer does a combinatorial search for known binding sites, specifically in Drosophila. It works quite well, I hear. Because comparative sequence analysis doesn't seem to work in flies, this is your best bet for cis-regulatory analysis.
MEME and MAST (http://meme.sdsc.edu/meme/intro.html)
MEME and MAST are programs to discover overrepresented motifs and search for known motifs, respectively. I haven't used them myself but they are standard tools that work fairly well. In particular, MEME is very sensitive albeit much slower than AlignACE.
PipMaker (http://pipmaker.bx.psu.edu/pipmaker/)
PipMaker does a local alignment of sequences using a BLAST algorithm and then displays them for you. You can compare multiple sequences with PipMaker. It works well.
VISTA (http://genome.lbl.gov/vista/)
VISTA is the most popular comparative sequence analysis program. It does a global alignment of medium-sized genomic regions and displays results where the similarity rises above a given threshold. It works well.
WormEnhancer (http://wormenhancer.org/Main)
WormEnhancer does a combinatorial search for known binding sites. It is the C. elegans version of FlyEnhancer, and it's the standard way to search for binding sites in worm.
YMF/Explanator (http://wingless.cs.washington.edu/YMF/YMFWeb/YMFInput.pl)
YMF/Explanator gives you a way to search for statistically overrepresented motifs, i.e. discover new binding sites. I've heard good things about it, although I have never used it myself.
Further Reading and References
(My apologies for the somewhat scattered selection. Even if the list isn't terribly comprehensive yet, all of these papers are worth reading!)
The de rigeur reference for the serious student of gene regulatory networks and regulatory regions:
The Regulatory Genome, by E.H. Davidson. ISBN 0120885638.
From DNA to Diversity is a less weighty introduction to the same topics:
From DNA to Diversity : Molecular Genetics and the Evolution of Animal Design, by Sean B. Carroll, Jennifer K. Grenier, and Scott D. Weatherbee. ISBN 1405119500.
The eve stripe 2 story is one of the earliest enhancer stories (and it's still one of the best-known):
Small S, Blair A, Levine M. Regulation of two pair-rule stripes by a single enhancer in the Drosophila embryo. Dev Biol. 1996 May 1;175(2):314-24. PMID: 8626035
The other original cis-regulatory story is that of endo16 in the sea urchin.
Yuh CH, Bolouri H, Davidson EH. Genomic cis-regulatory logic: experimental and computational analysis of a sea urchin gene. Science. 1998 Mar 20;279(5358):1896-902. PMID: 9506933
A paper demonstrating that comparative sequence analysis works well in sea urchins:
Yuh CH, Brown CT, Livi CB, Rowen L, Clarke PJ, Davidson EH. Patchy interspecific sequence similarities efficiently identify positive cis-regulatory elements in the sea urchin. Dev Biol. 2002 Jun 1;246(1):148-61. PMID: 12027440
An excellent paper that uses chick/human comparisons to find sox enhancers:
Uchikawa M, Ishida Y, Takemoto T, Kamachi Y, Kondoh H. Functional analysis of chicken Sox2 enhancers highlights an array of diverse regulatory elements that are conserved in mammals. Dev Cell. 2003 Apr;4(4):509-19. PMID: 12689590
This is the paper by Andy Cameron et al. that I referenced in the section on comparative sequence analysis.
Cameron RA, Chow SH, Berney K, Chiu TY, Yuan QA, Kramer A, Helguero A, Ransick A, Yun M, Davidson EH. An evolutionary constraint: strongly disfavored class of change in DNA sequence during divergence of cis-regulatory modules. Proc Natl Acad Sci U S A. 2005 Aug 16;102(33):11769-74. Epub 2005 Aug 8. PMID: 16087870
Here's a good paper on combinatorial pattern search in Drosophila:
Markstein M, Zinzen R, Markstein P, Yee KP, Erives A, Stathopoulos A, Levine M. A regulatory code for neurogenic gene expression in the Drosophila embryo. Development. 2004 May;131(10):2387-94. PMID: 15128669
Compensatory site gain & loss in Drosophilids:
Ludwig MZ, Palsson A, Alekseeva E, Bergman CM, Nathan J, Kreitman M. Functional evolution of a cis-regulatory module. PLoS Biol. 2005 Apr;3(4):e93. Epub 2005 Mar 15. PMID: 15757364
This is one the most beautiful recent papers on genome-wide cis-regulatory analyses; the authors analyzed a single interneuron-specific gene battery in worm.
Wenick, A.S. and Hobert, O. (2004). Genomic cis-regulatory architecture and trans-acting regulators of a single interneuron-specific gene battery in C. elegans. Dev. Cell 6, 757-770. [PMID: 15177025]
Here's a relatively recent review by the JASPAR folks:
Wasserman WW, Sandelin A. Applied bioinformatics for the identification of regulatory elements. Nat Rev Genet. 2004 Apr;5(4):276-87. Review. No abstract available. PMID: 15131651
Assessing binding site discovery tools:
Tompa M, Li N, Bailey TL, Church GM, De Moor B, Eskin E, Favorov AV, Frith MC, Fu Y, Kent WJ, Makeev VJ, Mironov AA, Noble WS, Pavesi G, Pesole G, Regnier M, Simonis N, Sinha S, Thijs G, van Helden J, Vandenbogaert M, Weng Z, Workman C, Ye C, Zhu Z. Assessing computational tools for the discovery of transcription factor binding sites. Nat Biotechnol. 2005 Jan;23(1):137-44. PMID: 15637633
Acknowledgements
A number of people read this over and critiqued it for me: in particular, Robert Zinzen, Erich Schwarz, Ali Mortazavi, and Alok Saldanha all offered trenchant comments that dramatically improved the quality of this screed.
Legalese
This document is Copyright (C) 2006, C. Titus Brown, titus@caltech.edu. Redistribution for fun (not profit!) is permitted as long as you keep this copyright notice attached.